Handbook |
Search Handbook
|
Barcode Data Validation
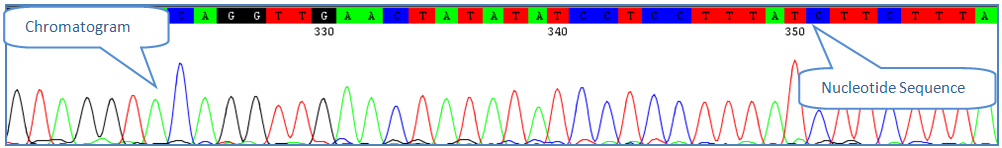
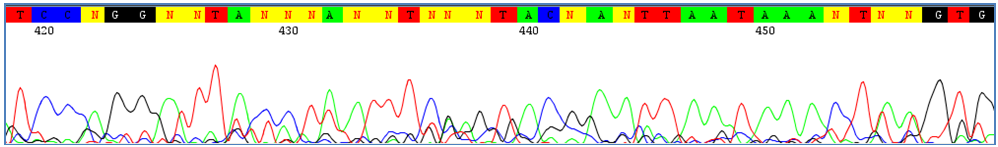
Sequence Assembly and AlignmentThe first step in validating the quality of the Barcode data in a project is certifying that the sequences have been properly assembled, edited, and aligned. In order to do that, users will need to have some basic knowledge about sequence editing and what to look for when editing sequences. This document provides only basic information on the process of sequence editing - for an advanced understanding on how to properly assemble and edit DNA sequences, using more specific sources is recommended. A trace file, also called a chromatogram, is a combination of a graphical representation of a DNA sequence and the matching nucleotide interpretation. Each chromatogram is composed of colour-coded peaks with each colour corresponding to one nucleotide. They are automatically supplied by DNA sequencing programs. In order to ensure the accuracy of sequences, it is important to manually check the quality of the traces. Errors are commonly found at the beginning and the end of the trace file where the signal intensity is weakest. Common Sequence Editing IssuesUnder the right conditions, trace files should require very little manual editing. However optimal conditions cannot always be met, and several issues can arise that may need to be corrected. 1) Minimal Background Noise
Trace files with minimal background noise may need more manual editing than high quality traces, but should still produce a reliable read. 2) Dye Blobs
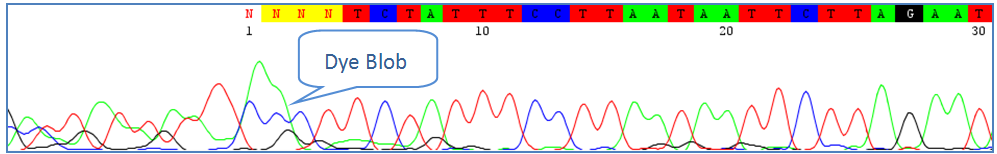
If the dye blob occurs at the beginning of the trace, it can be corrected by deleting the nucleotide sequence before the blob. If it occurs in the middle of the trace, it is best to leave the nucleotide sequence ambiguous. If sequencing was performed bidirectionally, the opposite trace might be able to rescue the final sequence. 3) Low Quality Traces
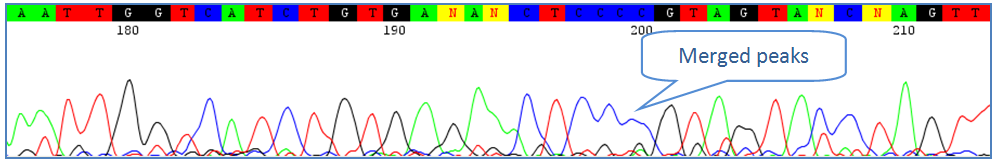
Low quality traces have peaks that are not well defined and contain a run of repeated bases that may appear to merge. These types of traces will require significant manual editing. Align to related sequences if possible to resolve repeat bases. 4) Partial Co-amplification of Contaminants
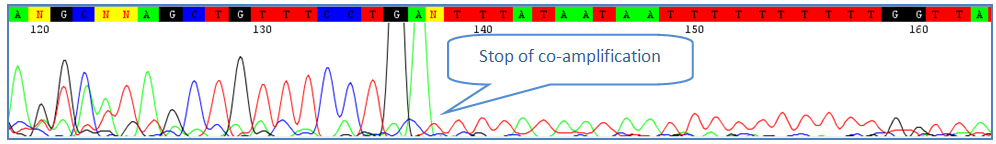
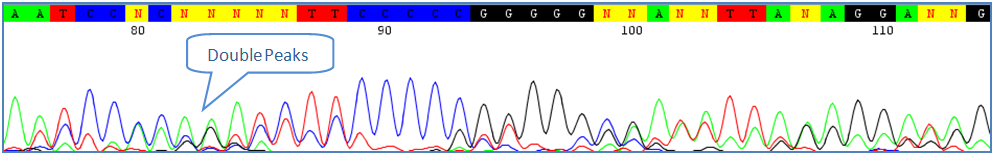
Sometimes only part of contaminating sequences gets amplified. This can be corrected by deleting the sequence upstream to the "drop" in signal. 5) Double-peaks in Co-amplification of Similar Sequences
Co-amplification is when two or more sequences from related species are amplified simultaneously. If only a few double-peaks are present in the Barcode region, they can be left as ambiguous bases at the discretion of the user. 6) Homopolimeric Tracts
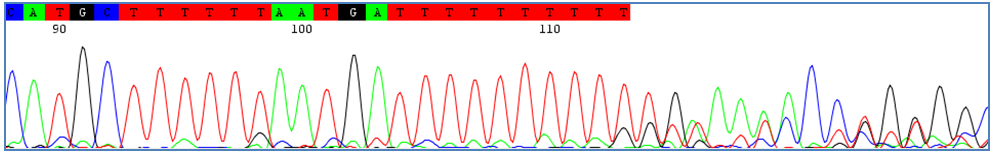
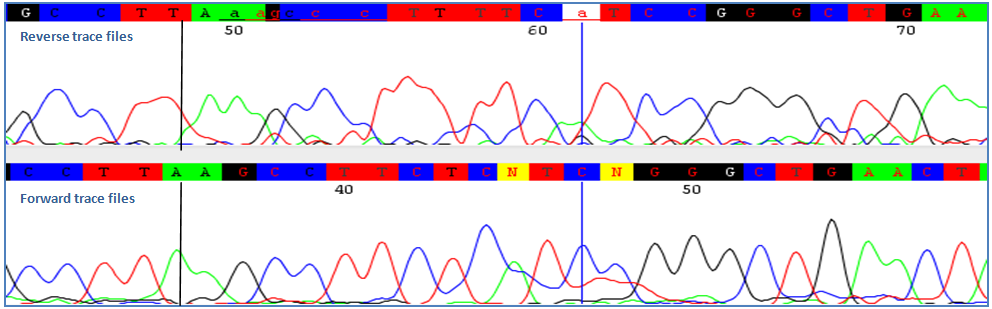
Homopolimeric tracts, or repetition of bases or sequences, are often natural occurrences and cannot be avoided. In certain situations, these tracts can result in out of sync sequences downstream from the tract. Traces with these tracts can often be rescued by bidirectional sequencing. To do this, delete the sequence downstream of the homopolymeric tract and align forward and reverse traces with a reference sequence. Manually overlap forward and reverse traces at the homopolymeric tract. 7) Alignment Errors
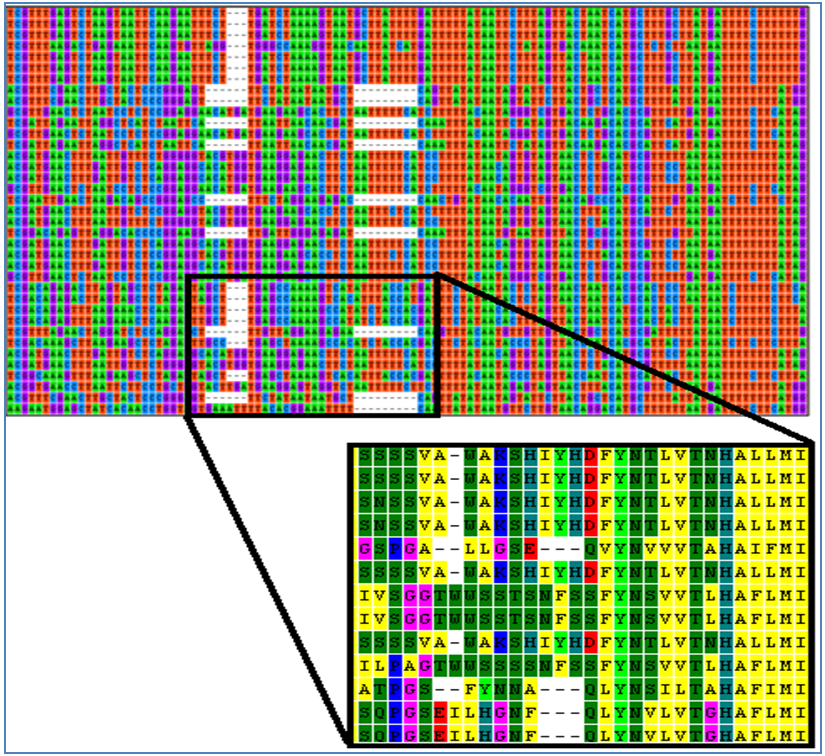
Shifts in the reading frame usually means a nucleotide or gap was added to one of the traces incorrectly. This type of error can usually be corrected by inspecting the properly aligned section of the sequence to determine the location of the mistake. 8) Indels (Insertion/Deletions)
Indels are insertions or deletions of nucleotides in a sequence. They can occur naturally during the evolutionary life of a species or as a result of poor sequence alignment. Identifying indels often requires comparing sequences from multiple species to determine where gaps should be located. Indels are naturally occurring and correctly placed if two criteria are met:
Animal groups with known indels:
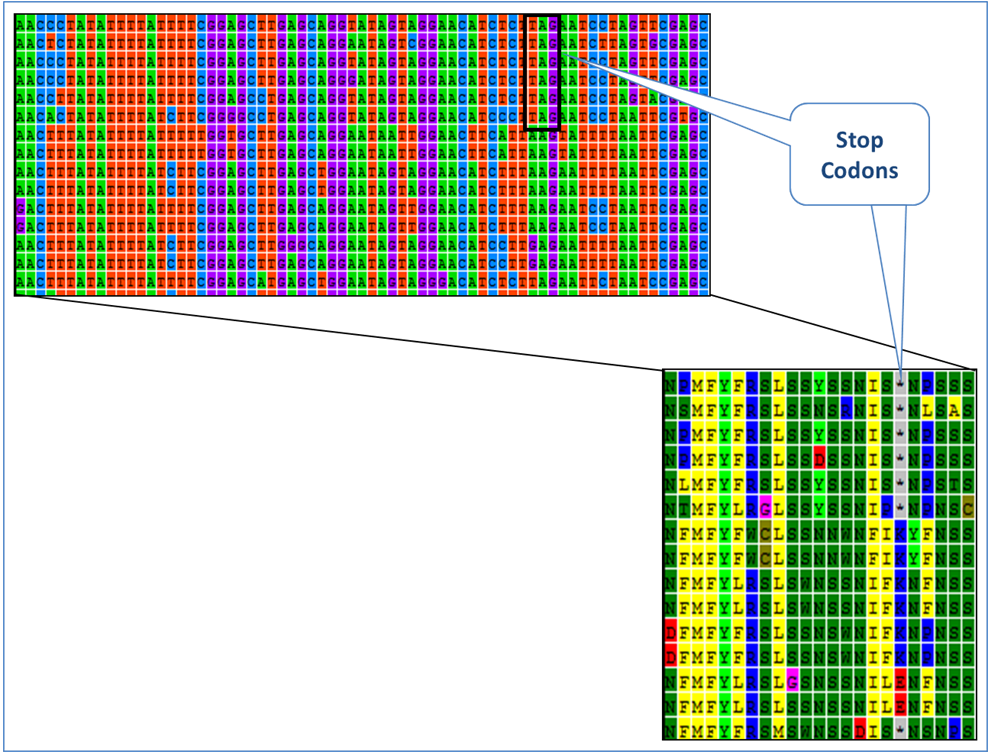
9) Stop Codons
10) PCR Primers included in the SequencePCR primers need to be removed from the sequence whenever possible to ensure the proper sequence length and reading frame are achieved. Different primers will be used depending on the taxonomic group being analyzed, so maintaining a copy of the primer sequence is essential to recognize and delete it from the sequence. The standard barcode length for most animal species is 658bp for sequences with no indels (see section 8. Indels). As long as traces contain approximately 500 bp of high quality sequence, PCR primers should be visible at the 3' end of each trace. Tips and TroubleshootingSometimes it is not possible to recognize the PCR primer in a trace. To ensure a trace file is trimmed in the correct nucleotide position, a sequence with the correct length from a closely related species can be downloaded from BOLD and aligned to the original trace. Using the BOLD sequence as reference, the trace can be trimmed to the same starting and ending point. This is an easy way to ensure trace files are in the correct reading frame.11) Reading Frame Shifts
The reading frame refers to the way a nucleotide sequence is translated into amino acids. There are 3 possible reading frames for a sequence, though only one is correct. A sequence is in the correct reading frame if translation starting at the second nucleotide results in a sequence with no stop codons. Tips and TroubleshootingBefore translating a sequence into amino acids, it is important to ensure that the correct genetic code table is being used. Most invertebrates will use a generic "invertebrate mitochondrial" translation table, however vertebrates and plants have their own specific tables. If the wrong table is used, false stop codons may appear in the sequence. Back to Top Trace File FailuresThe quality of trace files can vary considerably, but most traces will contain both high and low quality sections. Deciding when a trace file should be discarded can be a difficult decision. The following cases highlight when it is appropriate to discard a trace file. Case 1: Poor Quality DNA
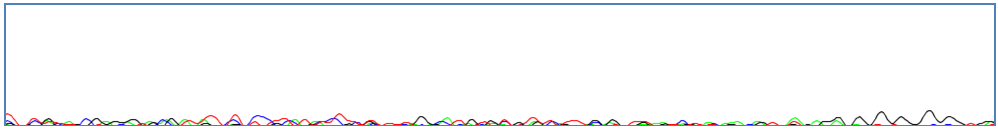
When the DNA used to generate a trace file is low quality, the peaks are often too ambiguous to call bases and the sequence cannot be corrected. Case 2: Complete Lack of DNA Amplification
Complete lack of amplification can occur when there is no PCR product in the sequencing reaction or the sequence reaction failed. Case 3: Contamination and Co-amplification of Unrelated Sequences
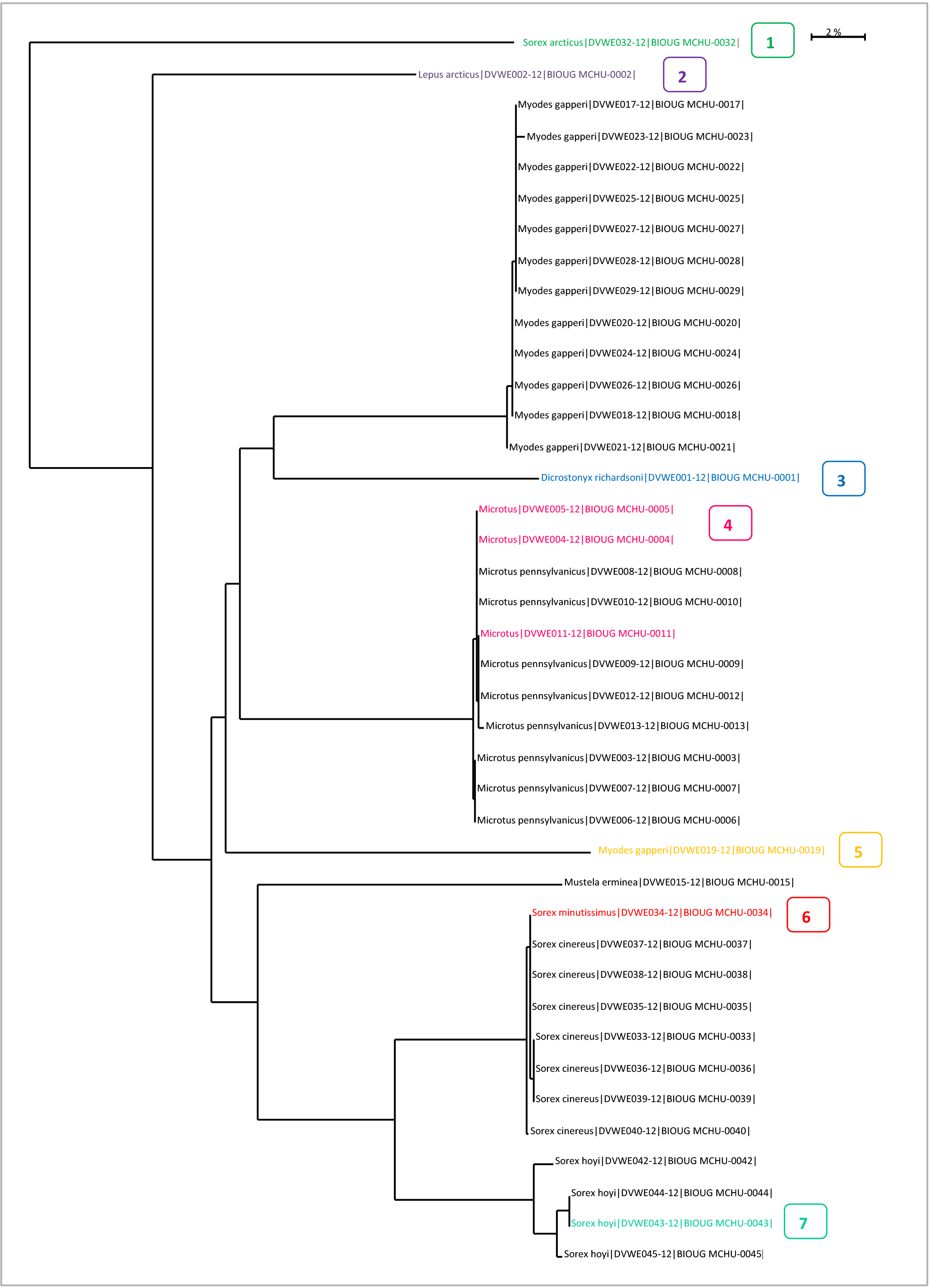
If two or more sequences from unrelated species are co-amplified, double-peaks will be present at almost every base position. Back to Top Taxon ID Trees for ValidationThe Taxon ID Tree on BOLD is a useful tool to identify problem sequences. Seven cases are described below.
Case 1: False outgroup resulted from a contaminationAn outgroup may be caused by contamination or it may be a real phenomenon resulting from a genetically distant taxa. The only way to know if an outgroup is the result of a contaminant is by comparing the nucleotide sequence to the BOLD ID engine database. To run a sequence against the BOLD ID engine:
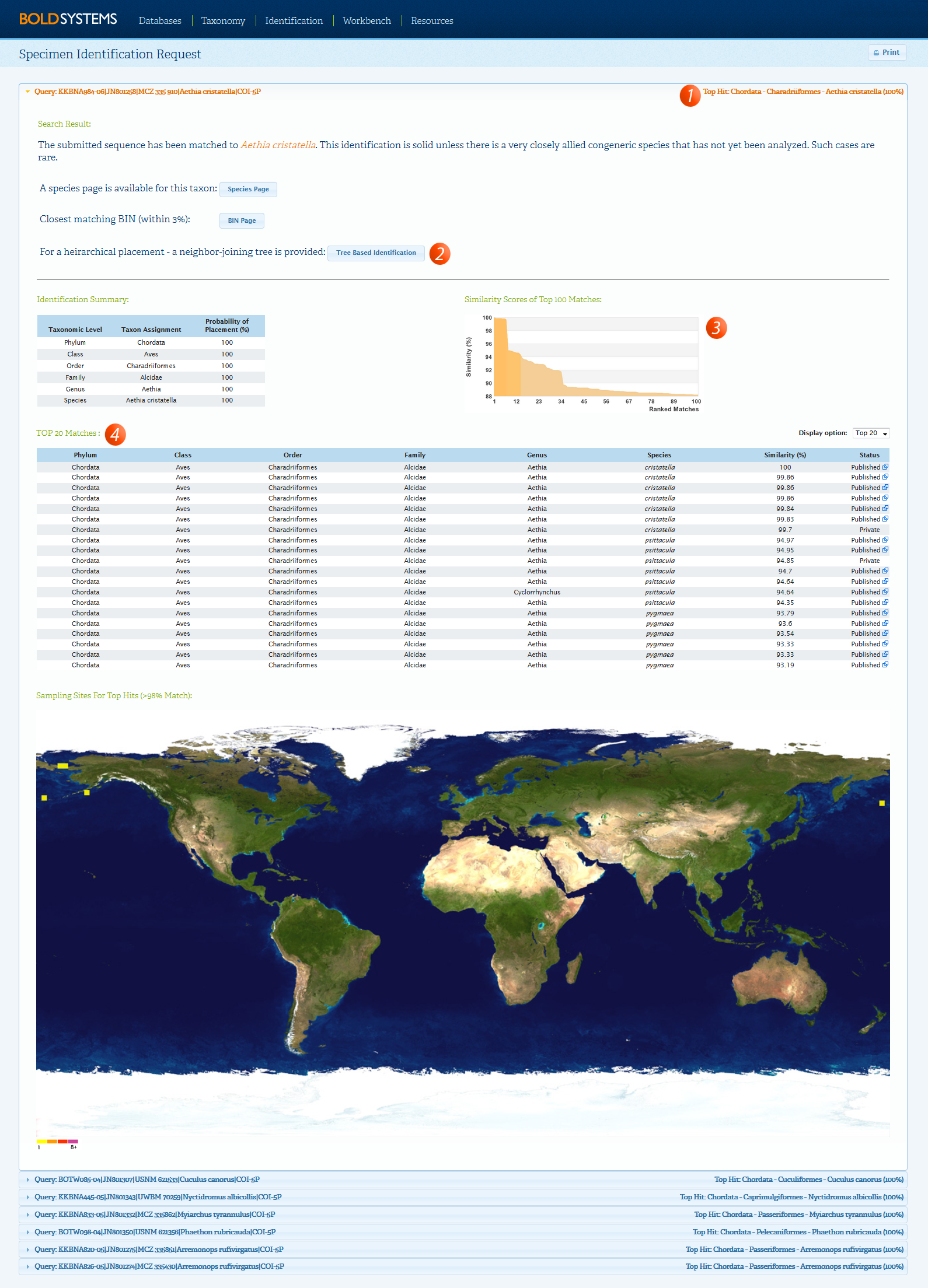
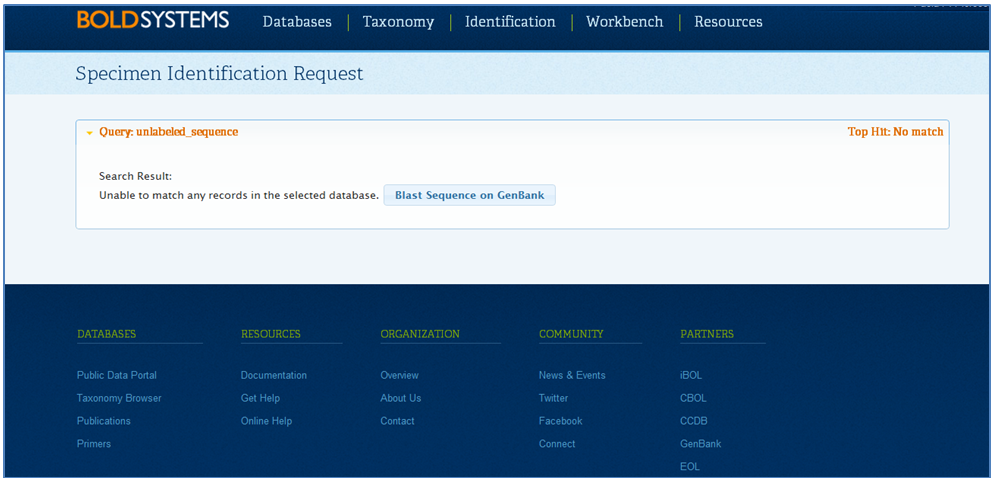
The Specimen Identification Request window will appear illustrating the top similarity matches as illustrated below. When the top match is at 99% similarity or higher and it does not agree with the taxonomic name provided, it usually indicates a contamination.
In this case, users can add annotation to the record to indicate the possible presence of a contamination. To add annotation to the affected records:
Case 2: Real outgroup resulted from a genetically unrelated taxonReal outgroups can sometimes be included on a tree. In order to determine if an outgroup is real or a contaminant, the sequence needs to be blasted against the BOLD ID Engine (refer to Case 1 - False outgroup resulted from a contamination for instructions on how to access the Identification Engine). If the outgroup represents a species new to BOLD, no record match will be displayed. The records should then be blasted against GenBank, which can be done directly from BOLD. When the BOLD ID Engine fails to find a match, click Blast Sequence on GenBank to directly access the Standard Nucleotide BLAST on GenBank. If the resulting identification on GenBank matches the name provided in the tree by more than 99%, it can be concluded that the identification is correct. This is a real outgroup and does not need to be tagged.
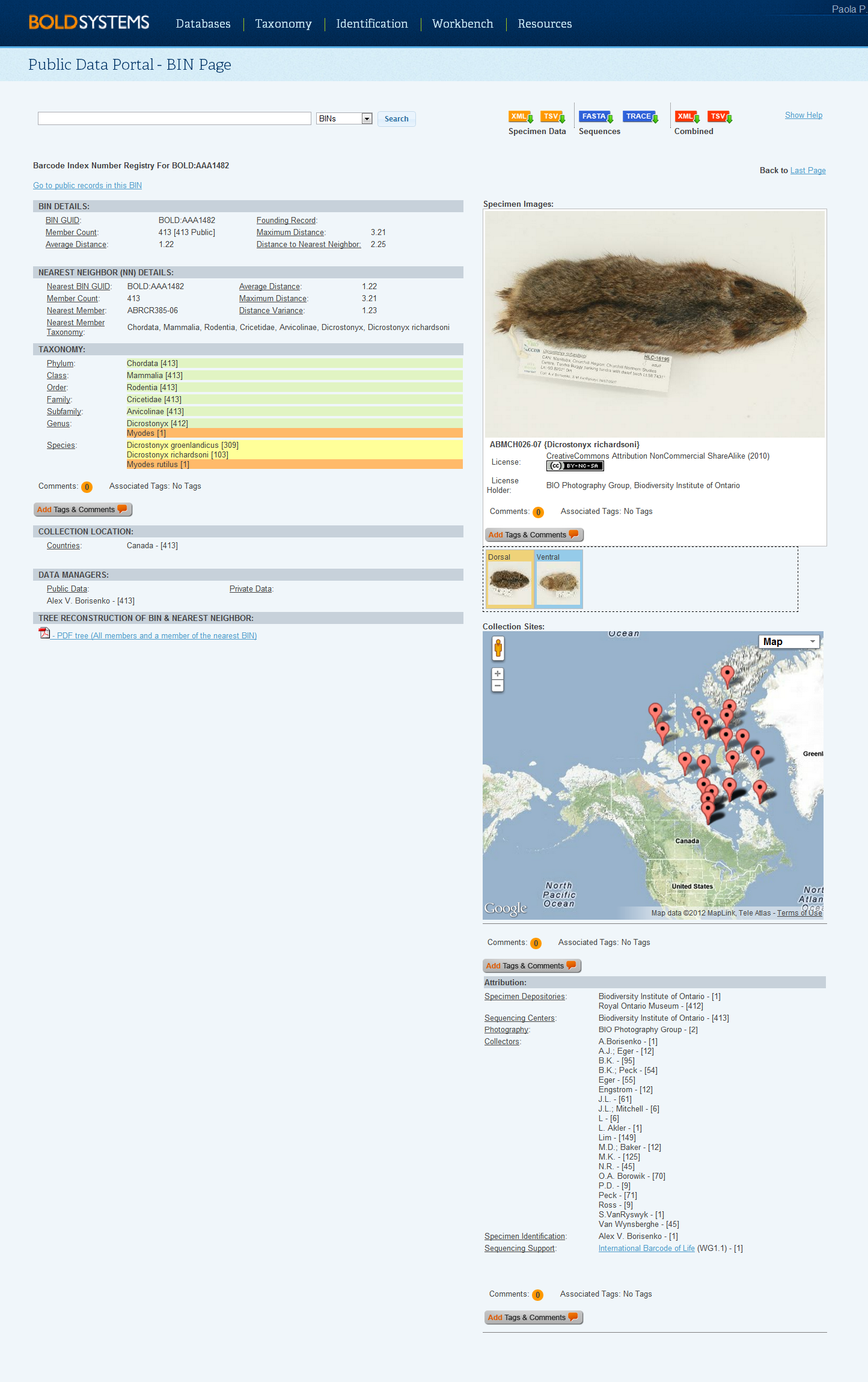
Case 3: Single branch resulted from unique recordSome species or haplotypes may appear as a single branch on the tree. It is important to check the identification of all single branches in a tree since these cannot be compared with other records within the same cluster. The Barcode Index Numbers (BIN) database can be used to confirm an identification; if the sequence meets the requirements to be clustered into BINs, then the record will have a BIN number. Refer to the Barcode Index Numbers (BIN) section in the Handbook. To navigate to the BIN page:
The data provided on the BIN page may help confirm the identity of single branch records on the tree, if other members of that species appear in other projects on BOLD. Where the correct identity of a single branch records cannot be confirmed right away, it is suggested that users monitor the BIN page for records over time as new specimens are being added to BOLD continously and activity on a BIN page is fluid.
Case 4: Incomplete identification on a clusterSome clusters on the tree may contain records that are identified to species and records that are not. It is possible to add full taxonomy to these records based on the tree and BOLD ID engine by sending a taxonomy update through the BOLD Submission Protocol. Tips and TroubleshootingWhen updating the taxonomy of a record based on the results from the identification engine, the Identified By field should be updated to "BOLD ID Engine". This informs other users that the identification provided was based on the record's nucleotide sequence without further examination of the voucher specimen and it should be reviewed by a taxonomic expert when possible. Further notes about taxonomic identifications can be added to the Taxonomy Notes and Identification Method fields. Case 5: Single branch resulted from contamination or misidentificationWhen two or more records with the same species name appear on a tree in separate branches, it is often the result of a contamination or misidentification. If a misidentification can be concluded and the correct identification is known, it is recommended that the taxonomy be updated as soon as possible without tagging the record. If a misidentification is not certain or the correct name is unknown, the record should be tagged and re-examined in the future. How to access record annotation:
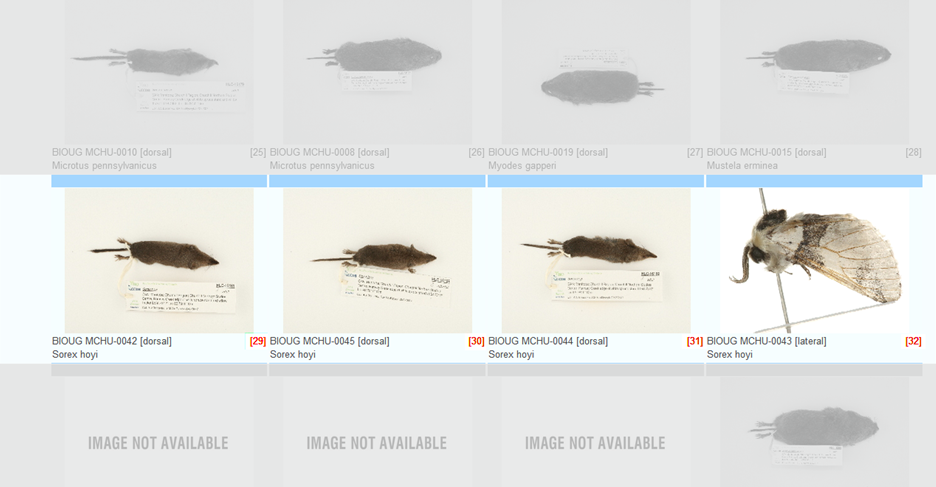
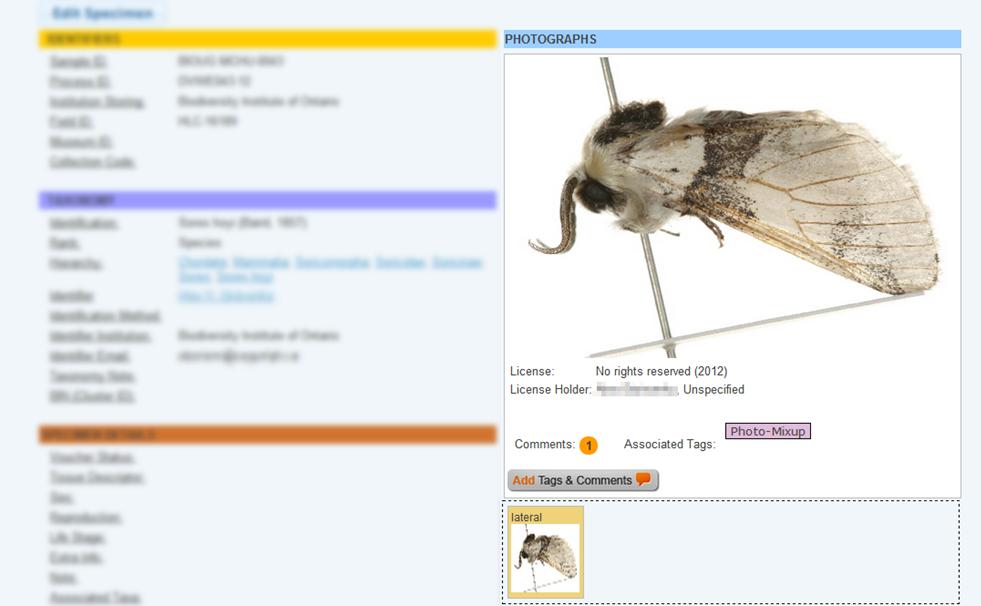
Case 6: Misidentified record in a cluster Some species can be difficult to identify solely on morphological characteristics. Sometimes Taxon ID trees can cluster together records that were believed to belong to two or more species. In certain cases this can be easily resolved by updating the taxonomy of misidentified records. Refer to the section on Updating Specimen Data. Tips and TroubleshootingBefore updating the taxonomy of any record in a project is important to check the sequence against the Identification Engine or BIN records (refer to the section on Identification Engine and BINs in this handbook) to ensure that the correct nomenclature matches other records on BOLD. Case 7: Image mismatchA mismatched image occurs when an incorrect picture is associated with a record. It is recommended to always create a matching image library when building a tree to examine records for this possible issue. Refer to the section on Taxon ID Trees in this handbook for more instructions on how to build a tree with matching images. When building the tree, choose "Matching Images and Spreadsheet" in the parameters window. Then from the Tree Result window choose the option to View Image List. Each branch on the tree will be automatically assigned a number that will correspond to a photo in the image library. See the screenshots below for an illustration.
To correct an image mismatch:
If the image mismatch cannot be resolved immediately, add a tag to the image to inform other users that this issue has been acknowledged. To add a tag on an image:
Back to Top |